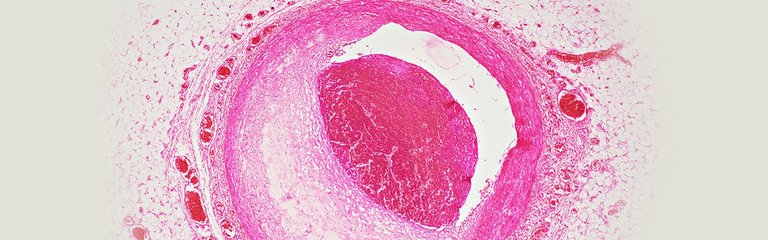
LDL-Cholesterin – eine der wichtigsten Ursachen für kardiovaskuläre Erkrankungen
Das Low-density-Lipoprotein (LDL-C) spielt eine Schlüsselrolle bei der Entstehung der Atherogenese und ist maßgeblich für die Entstehung atherosklerotischer kardiovaskulärer Erkrankungen, wie Myokardinfarkt, Angina Pectoris oder Schlaganfall verantwortlich.1,2
LDL-C – treibende Kraft der Atherogenese
Als Atherogenese – die Pathogenese der Atherosklerose – wird ein chronischer Entzündungsprozess von Gefäßen verstanden, der durch eine endotheliale Dysfunktion und u. a. einer Akkumulation bzw. Retention von u. a. LDL bzw. Cholesterin (Cholesterin-Estern) und Aktivierung von Immunzellen charakterisiert ist. Bereits im Kindesalter kann es zu einer beginnenden Lipidablagerung in der arteriellen Gefäßwand kommen. Frühe Stadien der Atherosklerose sind durch eine Dysfunktion des Endothels charakterisiert und werden durch zahlreiche kardiovaskuläre Risikofaktoren begünstigt.Der Prozess kann zu atherosklerotischen Plaques und – häufig erst nach Jahrzehnten – zu klinisch relevanten Komplikationen führen.1-3
Atherogenese – ein chronischer Entzündungsprozess

Retention und Modifikation von Lipoproteinen, Endothelschädigung
Initial kommt es u. a. bedingt durch die erhöhte Permeabilität des Endothels zur Akkumulation von LDL-C im subendothelialen Raum sowie zur Aggregation und oxidativen Modifikation von Lipoproteinen, insbesondere von LDL-Partikeln.4
Noxen wie Dyslipidämie, Tabakrauchen, arterielle Hypertonie und Störungen der Mikrozirkulation können das Endothel schädigen. Dabei setzt es u. a. vaskuläre (VCAM-1) und interzelluläre (ICAM-1) Adhäsionsmoleküle, Endothelin, Angiotensin II und gerinnungsaktive Substanzen frei.4
Makrophagen und Schaumzellentransformation
Als Folge der Endothelschädigung adhärieren Leukozyten leichter am Endothel und migrieren transendothelial. Die modifizierten Lipoproteine werden zunächst von gewebeständigen dendritischen Zellen und Makrophagen in der Intima aufgenommen; zusätzlich nehmen „patrouillierende“ Monozyten bereits frühzeitig im Verlauf der Atherogenese oxidiertes LDL über sog. Scavenger-Rezeptoren auf.
Die für die atherosklerotische Läsion charakteristischen Schaumzellen entstehen durch Differenzierung von Monozyten in Makrophagen im subendothelialen Raum. Sie „umfließen“ modifiziertes Cholesterin, während sie überschüssiges Cholesterin in Form von Cholesterin-Estern in Lipidtropfen speichern.4
Bildung der atherosklerotischen Plaque
Im weiteren Verlauf der Plaquebildung akkumulieren Makrophagen und T-Zellen und es kommt zur Apoptose der Zellen. Zudem aktivieren modifiziertes Cholesterin, Zytokine und Wachstumsfaktoren glatte Muskelzellen. Diese können modifizierte Lipoproteine aufnehmen und einen Makrophagen-ähnlichen Phänotyp annehmen.4
Die ständige Beladung der Makrophagen mit Lipiden geht mit endoplasmatischem Retikulumstress, programmiertem Zelltod (Apoptose) und programmierter Nekrose (Nekroptose) einher; Zelltrümmer werden unzureichend beseitigt. Es bildet sich ein nekrotischer Lipidkern, die nekrotische Läsion und das Entzündungsgeschehen nehmen zu.4
Plaqueruptur
Durch allmähliche Verdünnung der fibrösen Kappe wird die atherosklerotische Plaque schließlich zunehmend instabil. Diese kann aufgrund von Scherkräften rupturieren, so dass thrombogenes Material aus der Plaque freigesetzt wird. Das Gerinnungssystem wird aktiviert und es bilden sich nicht-okkludierende Thromben.4
Referenzen:
-
Beatriz HF et al. Immunobiology of atherosclerosis: a complex net of interactions. Int J Mol Sci 2019;20:5293.
-
Borén J et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2020;0:1-28.
-
Ference BA et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017;38:2459-2472.
-
Raggi P et al. Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions. Atherosclerosis 2018;276:98-108.