Familiäre Hypercholesterinämie – eine oft unerkannte Erkrankung
Die familiäre Hypercholesterinämie (FH) ist eine genetische, autosomal-dominant vererbbare Fettstoffwechselerkrankung, die durch eine ausgeprägte Erhöhung des LDL-C im Blutplasma charakterisiert ist.1-3 Die Entdeckung des LDL-Rezeptors und die Forschungsgrundlagen für die Entwicklung der HMG-CoA-Reduktasehemmer sind auf die Erforschung der familiären Hypercholesterinämie zurückzuführen.
Patient:innen mit einer diagnostizierten FH werden automatisch einer kardiovaskulären Hochrisikokategorie zugeordnet. Der Kategorie „hohes Risiko“ wird die FH ohne weitere Risikofaktoren zugeordnet. Sobald Patient:innen mit familiärer Hypercholesterinämie gleichzeitig einen prominenten Risikofaktor aufweisen, besteht ein „sehr hohes kardiovaskuläres Risiko“.3
Mithilfe einer auf den Patient:innen zugeschnittenen lipidsenkenden Therapie kann das kardiovaskuläre Risiko wirksam gesenkt werden.1,2
-

LDL-C-Zielwerte – je niedriger, desto besser
Die LDL-C-Zielwerte zur Reduktion des Risikos für kardiovaskuläre Ereignisse
orientieren sich an definierten Risikogruppen. Die Einteilung der Patient:innen
erfolgt anhand von bestehenden kardiovaskulären Erkrankungen, Risikofaktoren,
Vorliegen einer familiären Hypercholesterinämie und dem 10-Jahres-Risiko für eine
tödliche kardiovaskuläre Erkrankung (SCORE-System).1
Epidemiologie
- Mit einer geschätzten Prävalenz von 1: 200 bis 500 gehört die heterozygote familiäre Hypercholesterinämie (HeFH) zu den häufigsten genetischen Störungen.1
- Die Prävalenz der homozygoten familiären Hypercholesterinämie (HoFH) liegt bei 1: 300.000 bis 1.000.000 und ist aufgrund des Defekts auf beiden Allelen häufig mit einer schlechteren Prognose assoziiert.1
- In Deutschland wird die familiäre Hypercholesterinämie nur bei ca. 15 % der Patient:innen frühzeitig diagnostiziert, meist erst nach einem kardiovaskulären Ereignis, wie beispielsweise einem Herzinfarkt im jungen Alter bzw. bei familiärer Häufung von Myokardinfarkten.1
Pathogenese
Bei 85-90 % der Patient:innen liegt ursächlich eine Mutation im Gen des LDL-Rezeptors (LDL-R) vor.1 Aber auch genetische Defekte des Apolipoproteins B-100 oder eine „Gain-of-function“- Mutation der Protease PCSK9 kann zu einer Hypercholesterinämie führen.1,3
Folgen einer Funktionsstörung des LDL-R:
- Verminderte Aufnahme von LDL in die Leberzelle
- Gestörter LDL-Abbau
- Gesteigerte hepatische Cholesterinsynthese
- Vermehrte Bildung atherosklerotischer Plaques
- Extraplasmatische Cholesterinablagerungen
Klinisches Bild
Die familiäre Hypercholesterinämie wird in eine homozygote und heterozygote Manifestation unterteilt. Bei der häufigeren heterozygoten familiären Hypercholesterinämie ist die Aktivität der LDL-Rezeptoren um etwa 50 % vermindert. Eine supravalvuläre oder valvuläre Aortenstenose mit möglicher Einbeziehung der Koronarostien oder kutane gluteale Xanthome sind für Homozygotie typisch.1
|
|
Heterozygote Manifestation |
Homozygote Manifestation |
|
Prävalenz in Deutschland |
1 : 500 |
1 : 1.000.000 |
|
Typische LDL-C-Konzentrationen |
190–450 mg/dl |
> 400 mg/dl – 1.000 mg/dl |
|
Klinische Charakteristika |
Familiäre Manifestation |
Familiäre Manifestation |
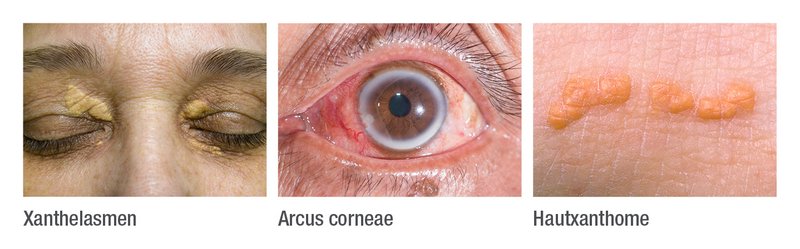
Diagnose der familiären Hypercholesterinämie
Die rechtzeitige Erkennung der familiären Hypercholesterinämie und eine effektive Therapie sind für den weiteren Verlauf und die Prognose entscheidend. Die klinische Diagnose beruht dem FH-Score des Dutch Lipid Clinic Networks zufolge auf der Beurteilung folgender Kriterien:4
- Positive Familienanamnese bei Angehörigen ersten Grades bzgl. KHK, vaskulärer Erkrankungen oder stark erhöhten LDL-C-Werten
- Anamnese mit vorzeitiger (Frauen < 60 Jahre, Männer < 55 Jahre) KHK, zerebrovaskulärer oder peripher-vaskulärer Erkrankung
- Nachweis extraplasmatischer Cholesterinablagerungen (Sehnenxanthome, Arcus corneae
- Erhöhten LDL-C-Spiegeln ohne lipidsenkende Therapie
- Molekulargenetischer Befund
Da das LDL-C bereits häufig vor Diagnosestellung mit einer Statintherapie gesenkt wurde, ist die Anamnese bezüglich familiärer kardiovaskulärer Ereignisse besonders wichtig.

Aufgrund des dominanten Erbgangs sollten neben den Indexpatient:innen auch Verwandte ersten Grades molekulargenetisch untersucht werden. In die molekulargenetische Analyse werden insbesondere folgende Gene einbezogen:3
- LDL-R
- Apolipoprotein B-100 (apoB) (rezeptorbindende Domäne)
- PCSK9 (proprotein convertase subtilisin/kexin type 9)
- Ggf. LDLRAP1 (low density lipoprotein receptor adaptor protein 1)
Der Nachweis einer funktionell relevanten Mutation beweist das Vorliegen einer familiären Hypercholesterinämie. Bei 80 % der Patient:innen mit Verdacht auf die Erkrankung (LDL-C, Familienanamnese) ist der Nachweis der relevanten Mutation nach systematischer klinischer Vorauswahl erfolgreich.1

Empfehlung: Die LDL-Senken Patient:innenseite
Auf der Patient:innenseite finden Sie informative Materialien, zum Beispiel Broschüren und Checklisten, für Ihre Patient:innen rund um das Thema LDL-Cholesterin.
Leitliniengerechte Therapie der familiären Hypercholesterinämie
Eine adäquate cholesterinsenkende Therapie der Patient:innen ist mit einer klaren Verbesserung der Prognose assoziiert. Die ESC/EAS-Leitlinie empfiehlt als therapeutischen Standard bei erhöhten LDL-C-Werten eine möglichst frühzeitige Behandlung mit einem Statin. Bei Kindern vor dem 10. Lebensjahr werden nicht medikamentöse sowie diätetische Therapieoptionen vorgeschlagen.1,3

Für Ärzt:innen:
Kostenfreier Zugriff auf alle Inhalte mit Ihrem DocCheck-LogIn
Mit Ihrem DocCheck-Login erhalten Sie freien Zugang zu allen, auch den geschützten, Inhalten, Grafiken, Fortbildungen und Artikeln auf dieser Seite.
Sie haben noch kein DocCheck-LogIn, oder haben Ihre Zugangsadaten nicht zur Hand, klicken Sie für Registrierung oder zum Erhalt Ihrer Zugangsdaten einfach auf die Links "Passwort vergessen" oder "Registrieren" im LogIn Fenster.
Aus gesetzlichen Gründen (Heilmittelwerbegesetz, § 10, Abs. 1) dürfen wir Patient:innen leider nicht über verschreibungspflichtige Medikamente informieren. Wir wünschen uns gut informierte Patient:innen. Wir bedauern daher, dass wir wegen dieser Rechtslage den Patient:innen unsere Informationen nicht zur Verfügung stellen dürfen.


Kardiovaskuläres Risiko reduzieren – mit diesen Lipidsenkern
Zur Behandlung der Hypercholesterinämie stehen verschiedene Behandlungsansätze zur Verfügung. Lesen Sie von konservativen Ansätzen und den medikamentösen Optionen.

Infoblatt Patient:innen – alles über familiäre Hypercholesterinämie
Wichtige Daten und Fakten rund um die Erkrankung – als praktisches Handout für Ihre Patient:innen. Ganz einfach hier downloaden oder direkt bestellen.

Lipidtherapie: mit der richtigen Kombination zum Erfolg
Wie lässt sich die lipidsenkende Therapie gestalten, damit mehr Patient:innen die strengen LDL-C-Zielwerte errichen können? Ein Einblick in aktuelle Studiendaten gibt Aufschluss.
Referenzen:
-
Klose G et al. Familiäre Hypercholesterinämie: Entwicklungen in Diagnostik und Behandlung. Dtsch Ärztebl Int 2014;111:523-529.
-
Diagnostik und Therapie bei familiärer Hypercholesterinämie. DACH-Gesellschaft Prävention von Herz-Kreislauf-Erkrankungen e.V.: https://www.dach-praevention.eu/fh-diagnostik-und-therapie/; abgerufen am 18.06.2020
-
Mach F et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur Heart J 2020; 41: 111-188.
-
Nordestgaard BG, Chapman JM, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Consensus Statement of the European Atherosclerosis Society. European Heart Journal 2013; 34:3478-3490.
-
Duell PB et al. Longitudinal low density lipoprotein cholesterol goal achievement and cardiovascular outcomes among adult patients with familial hypercholesterolemia: The CASCADE FH registry. Atherosclerosis 2019;289:85-93.